

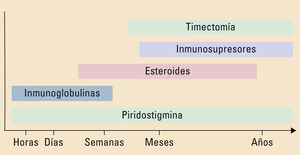


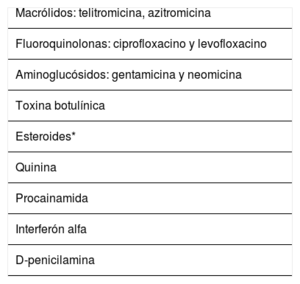

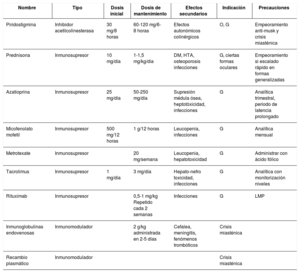
La miastenia gravis (MG) es la enfermedad de la unión neuromuscular más frecuente. Se trata de un trastorno adquirido de base autoinmune, en el que se detectan diferentes anticuerpos contra elementos del receptor muscular postsináptico. Los síntomas iniciales suelen aparecer en un grupo muscular aislado, preferentemente en la musculatura ocular con aparición de ptosis y diplopía. Posteriormente, los síntomas progresan con afectación muscular generalizada, apareciendo una afectación bulbar con síntomas de disfagia y disartria, debilidad de la musculatura cervical y a nivel proximal de las extremidades. Si la progresión clínica de la MG continúa, los pacientes desarrollan una crisis miasténica, asociándose fallo respiratorio. El rasgo clínico más característico de la MG es la variabilidad de la debilidad, con fluctuaciones clínicas dentro del mismo día, en relación con el ejercicio o factores agravantes, alcanzando en ocasiones una remisión espontánea. La sospecha clínica es clave tras reconocer este patrón clínico y confirmar el diagnóstico, principalmente con la determinación en sangre de los anticuerpos característicos de la MG acompañada del estudio neurofisiológico. El avance en el conocimiento de la inmunopatología subyacente ha permitido la optimización de las estrategias terapéuticas, combinando inhibidores de la acetilcolinesterasa, inmunosupresores y timectomía, con la consiguiente disminución en su morbimortalidad.
Palabras clave
Myasthenia gravis (MG) is the most common disease of the neuromuscular junction. It is an acquired autoimmune disease, in which several antibodies against postsynaptic receptors are detected. Initial symptoms often manifest focally, mainly ocular weakness resulting in ptosis and diplopia. Subsequently, symptomatology evolves to generalized muscular involvement and bulbar symptoms: dysphagia and dysarthria, cervical musculature and proximal limb weakness. Clinical progression results on myasthenic crisis and respiratory failure. The clinical hallmark of MG is a fluctuating pronounced muscular weakness along the same day and related with physical activity or aggravating factors. Occasionally, spontaneous remissions have been reported. Diagnosis key is clinical suspicion achieving by clinical pattern recognition. Diagnostic confirmation is obtained by serological (MG antibodies) and neurophysiological tests. Thanks to advances in the knowledge about underlying immunopathology, the range of therapeutic strategies has grown (combining acetylcholinesterase inhibitors with immunosuppressant drugs and thymectomy), decreasing its morbidity and mortality.
Keywords
Identifíquese
¿Aún no es suscriptor de la revista?

Comprar el acceso al artículo
Comprando el artículo el pdf del mismo podrá ser descargado
Precio: 19,34 €
Teléfono para incidencias
De lunes a viernes de 9h a 18h (GMT+1) excepto los meses de julio y agosto que será de 9 a 15h