
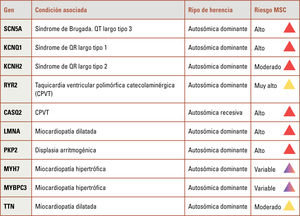
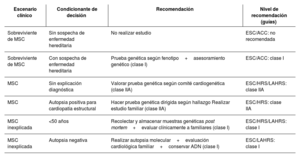
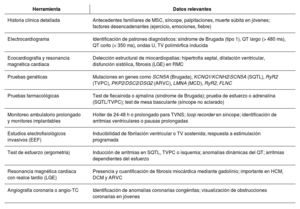
La muerte súbita cardíaca (MSC) representa un desafío clínico y forense de alto impacto. La integración de pruebas genéticas y autopsias moleculares ha revolucionado su abordaje, permitiendo identificar causas hereditarias incluso en ausencia de anomalías estructurales. Hasta un 40% de los casos sin causa evidente podrían explicarse por variantes genéticas patogénicas, lo que justifica la implementación de estrategias diagnósticas multidisciplinarias. La estandarización de protocolos para la recolección de ADN, la interpretación cuidadosa de variantes (patogénicas, benignas o inciertas) y la incorporación de estudios complementarios son pilares esenciales en la estratificación del riesgo. Enfermedades como canalopatías y miocardiopatías hereditarias explican una fracción significativa de los eventos, y su detección permite establecer medidas preventivas. A nivel epidemiológico, la MSC constituye entre el 10% y el 15% de las muertes naturales en países desarrollados. En contextos donde la autopsia es obligatoria, como en Reino Unido o Dinamarca, se han logrado mayores avances diagnósticos. El consentimiento informado y la regulación bioética de las pruebas genéticas post mortem son fundamentales para proteger la autonomía familiar y garantizar el uso adecuado de los resultados. Se recomienda un enfoque integral y dinámico, basado en evidencia científica y adaptado a los avances tecnológicos para mejorar el diagnóstico, el manejo y la prevención de la MSC en pacientes y familiares de riesgo.
Palabras clave
Sudden cardiac death (SCD) represents a high-impact clinical and forensic challenge. The integration of genetic testing and molecular autopsies has revolutionized the approach, making it possible to identify hereditary causes even in the absence of structural anomalies. Up to 40% of cases with no evident cause could be explained by pathogenic genetic variants, which justifies the implementation of multidisciplinary diagnostic strategies. The standardization of protocols for DNA collection, the careful interpretation of variants (pathogenic, benign, or uncertain), and the incorporation of additional studies are essential pillars in risk stratification. Diseases such as channelopathies and hereditary cardiomyopathies account for a significant fraction of the events and their detection allows for establishing preventive measures. Epidemiologically, SCD accounts for 10% to 15% of natural deaths in developed countries. In contexts where autopsy is mandatory, as in the United Kingdom or Denmark, greater diagnostic advances have been achieved. Informed consent and bioethical regulation of post-mortem genetic testing are essential to protect family autonomy and ensure the appropriate use of the results. A comprehensive, dynamic approach based on scientific evidence and adapted to technological advances is recommended to improve the diagnosis, management, and prevention of SCD in at-risk patients and family members.
Keywords
Identifíquese
¿Aún no es suscriptor de la revista?

Comprar el acceso al artículo
Comprando el artículo el pdf del mismo podrá ser descargado
Teléfono para incidencias
De lunes a viernes de 9h a 18h (GMT+1) excepto los meses de julio y agosto que será de 9 a 15h